Navigation auf uzh.ch
Navigation auf uzh.ch
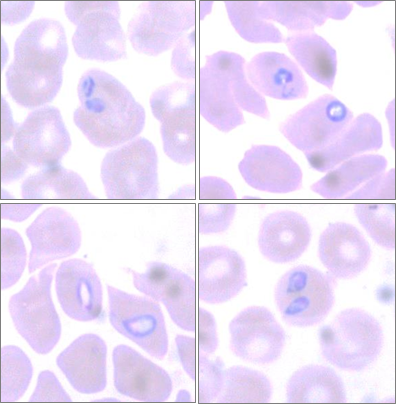
Canine babesiosis is caused by an apicomplexan parasite of the genus Babesia which is transmitted by ticks. Babesiosis is characterised by an intraerythrocytic parasitism which leads to severe clinical signs and often cause death if left untreated. Canine babesiosis is considered as an emerging vector-borne disease, as numerous cases have been reported in new areas throughout Europe, including focal regions in Switzerland (Eichenberger et al., 2015, Ticks Tick Borne Dis.). Currently, host-parasite interactions of this parasite are poorly understood. Hence, a significant improvement of our knowledge about the molecular details of the pathophysiology is required as a prerequisite for new intervention approaches. An early diagnosis of canine babesiosis increases the chance of survival. Correspondingly, a poor outcome in acute B. canis infection is indicated by changes in the laboratory profile (Eichenberger et al., 2015, J Vet Intern Med.). In practice, diagnosis of canine babesiosis is based on morphological and molecular appearance of the parasite. Aim of this project was the establishment of alternative diagnostic test assays using specific monoclonal antibodies against iEc membrane-associated parasite proteins or circulating parasite antigens. In this project we could confirm the potential of the detection of circulating Babesia-antigens in the blood of infected dogs with a sandwich ELISA (Eichenberger et al., 2017, Vet Parasitol.). This test will be further developed for the use in epidemiological investigations and for the establishment of a rapid diagnostic assay for the use in the practice.
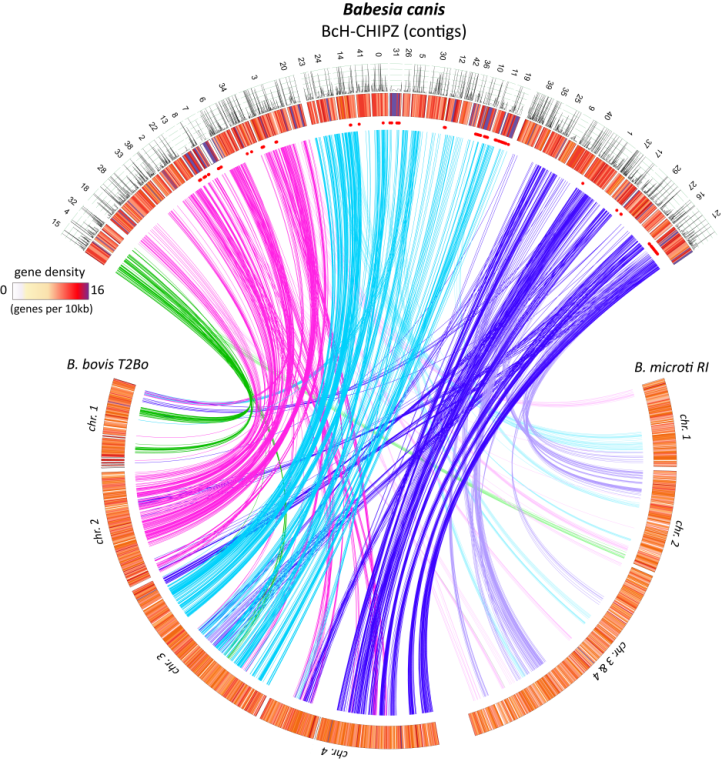
Another aim of the project was to identify and analyse the secreted B. canis proteome which affects iEc biology and host immunology. A prerequisite was the establishment of in vivo and in vitro models for canine babesiosis. This study leads to a model for the acute babesiosis in order to explore proteomic parasite-host interactions and improve diagnostics. We combined high-throughput approaches with a detailed characterization of these parasite determinants and their interactions with host receptors. As a backdrop, we generated a fully annotated B. canis genome sequence of a virulent field isolate underpinned by extensive genome-wide RNA-seq analysis. In silico analysis of the 3467 annotated gene models identified 509 potential secreted parasite proteins. By a shotgun proteomics approach of exported parasite factors, we found evidence for conserved factors in apicomplexan hemoparasites involved in immune evasion (e.g. VESA-protein family), proteins secreted across the iRBC membrane into the host bloodstream (e.g. SA- and Bc28 protein families), potential moonlighting proteins (e.g. profilin and histones), and uncharacterized antigens present during acute crisis in dogs (Eichenberger et al., 2017, Sci Rep.). This data contribute decisively to build an accurate model for parasite-host interaction, which can be exploited for innovative intervention strategies aimed at facilitating diagnosis and management of canine babesiosis.
Institute members: Ramon Eichenberger (PhD student), Štefanić Saša, Adrian Hehl, Peter Deplazes (Project Leader)
Funding sources: Forschungskredit der Universität Zürich
In collaboration with: Functional Genomics Center Zurich (FGCZ), Switzerland; University of Zurich (UZH), Switzerland; Alicia Rojas, Departamento de Parasitologia, Centro de Investigación Enfermedades Tropicales, Universidad de Costa Rica, Costa Rica; Torsten J. Naucke, Department of Zoology, Division of Parasitology, University of Hohenheim, Germany.
|
Stained preparation of dog erythrocytes containing |
Comparison of the assembled Babesia canis genometo related Babesia species based on synteny (conserved genes). Despite significant species-specific |